What is the cell cycle?
The cell cycle, also called the cell division cycle, refers to the process from the completion of the division of a continuously dividing cell to the completion of the next division. In this biochemical process, a large amount of DNA in a cell’s chromosome is precisely duplicated, and then the copies are split exactly into two genetically identical daughter cells.
The function of the cell cycle
The cell cycle Recombinants is an important process by which a single-celled fertilized egg develops into a mature organism. And the cell cycle maintains and ensures the regeneration of hair, skin, blood cells and some internal organs.
The cell cycle process
In eukaryotic cells or cells with a nucleus, the phases of the cell cycle include two main stages: the interphase and the mitotic (M) phase.
1. Interphase:
Interphase is the duration between the end of a cell’s last division and the beginning of its next division. Interphase is an important period during mitosis. And it prepares for cell division and makes the next mitosis possible. Many events take place during interphases, such as DNA replication, the synthesis of related proteins, and the gradual disappearance of the nuclear membrane and nucleoli. And interphase time accounts for about 91 per cent of cell division. Interphase comprises Gap 1 (G1) phase, Synthesis (S) phase, Gap 2 (G2) phase.
Sometimes, some cells drop out of the cycle and stop dividing under adverse conditions, such as nutrient deprivation. These cells enter a resting phase, the G0 phase. Among these cells, some could re-enter the cycle when given the right conditions, but some cells that do not proliferate can no longer divide like neuron cells: they have reached their final stage of development. So the cells that reside in the G0 phase can be a temporary or permanent rest.
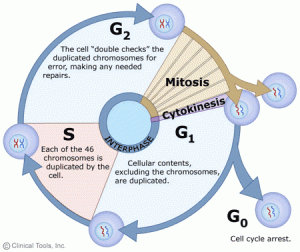
G1: Also called the growth phase. In the duration of the G1 phase, the organelles enlarge and mRNA and protein are synthesized in the cells. And the cells get bigger. All of these alterations are primed for DNA synthesis. The checkpoint (restriction point) in the G1 phase determines whether a cell continues to divide or exits the cycle (enters the G0 phase). If a cell successfully passes through the G1 checkpoint, it acquires the admission ticket to continue dividing. The checkpoint is regulated by cyclin G1/S, which promotes the transition of cells from the G1 to the S phase.
S: The S phase completes the synthesis of DNA and histones that are involved in DNA assembly and chromatin composition. During this period, the amount of DNA doubles and each chromosome is copied into two chromatids that are joined at the centromere.
G2: The G2 phase is the gap between the end of DNA replication and the beginning of mitosis. Cells synthesize certain proteins and RNA molecules to provide material to enter mitosis during the G2 phase. The G2 checkpoint, which is regulated primarily by the tumour protein p53, examines the cell for DNA damage within the chromosome before the cell enters the mitotic phase. Once it traces DNA damage, p53 can either repair DNA or trigger apoptosis. If p53 mutates or malfunctions, DNA-damaged cells can continue through the cell cycle, potentially leading to the development of cancer.
2. Mitotic Phase (M):
Mitosis occurs only in eukaryotes. Prokaryotes divide by binary division due to the absence of nuclei. The M phase is complex and highly regulated. The sequence of events is divided into prophase (including prophase and prometaphase), metaphase, anaphase, and telophase. Mitosis accounts for about 10 per cent of the cell cycle (it can last only an hour or two) and is much shorter than interphase. Mitotic errors can cause cell death by apoptosis or mutations that can induce cancer.
Prophase: During prophase, the nuclear envelope disintegrates and the nucleolus disappears. Chromatin condenses into coils to form chromosomes. And the centrosome emits star rays (in animal cells) or the cell poles send out spindle filaments (in plant cells) to form spindles. And then the spindle fibres attach to the centromere of the chromosome.
Metaphase: When the spindle fibres attach to the centromeres, they pull the chromosomes toward the centre of the cells, where all the chromosomes line up at the surface of the equator.
Anaphase: In this stage, the centromere splits and the two sister chromatids are attracted to two poles of the cells by the spindle fibre. As a result, the number of chromosomes doubles.
Telophase: the nuclear envelope reforms and the nucleolus appears. Also, the chromosomes gradually unzip to form chromatin, and the spindles gradually disappear. Subsequently, cytokinesis occurs, which divides the nucleus, cytoplasm, organelles, and cell membrane into two daughter cells that contain nearly equal parts of the parent cell.
Cytokinesis works differently in animal cells and plant cells. In animal cells, a protein near the equator forms the contraction ring to pinch the cell in half, creating shallow grooves on the surface. Due to cell walls, plant cells do not form shrinking rings but instead, build cell plates in the middle of the cells and then regenerate new cell walls in the two daughter cells.
Regulation of the Eukaryotic Cell Cycle
Regulation of the eukaryotic cell cycle, including detection and repair of DNA damage and prevention of uncontrolled cell division, is critical for cell survival. The molecular events that control the cell cycle are ordered and directed. The completion of the cell cycle process depends on the precise and strict regulation of the cell cycle by various regulatory factors. The core of these regulatory factors is:
Cyclin-dependent kinase (CDK): Cyclin-dependent kinase inhibitor (CKI) is a negative regulatory factor for CDK, while cyclin can upregulate CDK.
MPF (M phase promoting factor): It is a factor that can induce interphase cells to enter the division stage early in the M phase cells of all eukaryotes. MPF can catalyze the CDK subunit so that it remains constant in amount. And it is regulated by Cyclin. MFP accumulates and is broken down at different phases of the cell cycle.
Cellular Checkpoints: Primarily detect whether early cell cycle events have been completed and cells are intact, and monitor DNA damage or delay responses during cell cycle progression. Here are some cell checkpoints listed below.
● The G1/S checkpoint is the rate-limiting step in the cell cycle. It is responsible for checking if the cells have enough material to fully replicate the DNA (nucleotide bases, DNA synthase, chromatin, etc.).
● The S checkpoint checks whether DNA replication is complete.
● The G 2 / M checkpoint is the checkpoint where the cell makes sure it has enough cytoplasm and phospholipids to divide the cell in two. Sometimes it checks if the replication time is correct.
● The metaphase-anaphase checkpoint is the test point of the spindle assembly. Failure during the attachment of centromeres to the spindle inhibits APC activity, resulting in cell cycle arrest.
Cell Cycle and Diseases
The cell cycle is associated with a variety of human diseases, especially cancer. Uncontrolled cell proliferation caused by cell cycle disorders is the main cause of cancer. At the molecular level, it is the result of genetic mutations that cause inappropriate activation of cell cycle promoters and/or deactivation of inhibitors, resulting in uncontrolled cell cycle regulation.
Related Cell Cycle Applications
Recently, some experts have designed some drugs that aim to stop cell spindle formation and further inhibit cell mitosis and maintain cell division in the G0 phase. The drugs effectively slow down malignant proliferation and the spread of cancer cells.
For example, ordinary watermelons are diploid and produce normal seeds. Ordinary watermelons treated with colchicine can make tetraploid watermelons and produce normal seeds. Due to colchicine’s inhibition of spindle formation, mitosis is repressed and chromosomes are arrested at the metaphase of division. In such mitosis, the chromosomes divide longitudinally, but the cells do not divide and cannot form two daughter cells, so the chromosomes are duplicated. When a diploid watermelon is crossed with a tetraploid watermelon, the triploid watermelon is produced. Since triploid watermelon cannot distribute chromosomes equally among gametes, normal seeds cannot be obtained. This is a seedless watermelon